
Carbamyl Phosphate Synthetase I Deficiency (CPS I Deficiency)
This acute illness protocol is a guideline for healthcare professionals treating the sick infant or child who is known to have carbamyl phosphate synthetase I deficiency (CPS I deficiency). The protocol was developed at Boston Children’s Hospital under the direction of Dr. Harvey Levy, Senior Physician in Medicine/Genetics and Dr. Jonathan Picker, Fragile X Program Director, and was updated by Dr. Levy.
Disclaimer
Metabolic crises in infants and children with urea cycle disorders are complex medical emergencies and must be treated as such to avoid death or serious brain injury. This protocol is only a guideline and should not be used for definitive treatment without metabolic consultation. It is essential to call or page the on-call genetics/metabolism fellow, or failing this, the on-call metabolic attending at your hospital or nearest pediatric tertiary care center, as rapidly as possible. Please read our Terms of Use.
Pathophysiology
CPS I deficiency is one of the proximal urea cycle defects and is due to a complete or partial deficiency of the mitochondrial enzyme carbamyl phosphate synthetase I (CPS I) which produces carbamyl phosphate from ammonia, ATP, and HCO3 (as shown below). N-acetylglutamate (formed by the condensation of glutamate with acetyl CoA in the mitochondria catalyzed by N-acetylglutamate synthase (NAGS)) is an obligatory activator of the CPS I enzyme. CPS I deficiency is inherited in an autosomal recessive manner, thus it affects males and females equally.
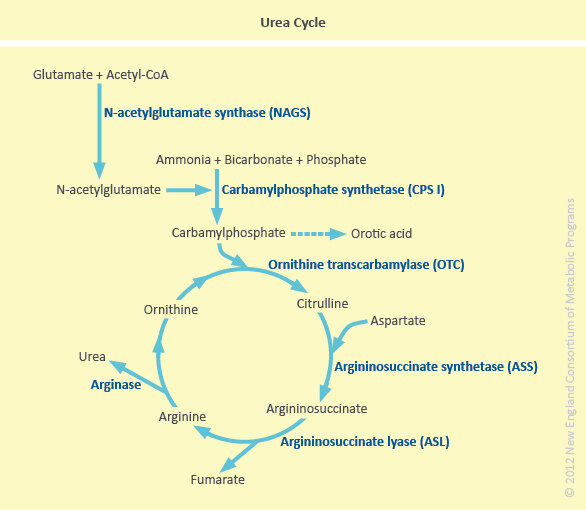
The liver is the only site of the complete urea cycle. Among the six enzymes in the cycle, N-acetylglutamate synthase (NAGS), carbamyl phosphate synthetase I (CPS I) and ornithine transcarbamylase (OTC) are intramitochondrial whereas arginase, argininosuccinate synthetase (ASS) and argininosuccinate lyase (ASL) are cytosolic. CPS I deficiency is one of the less frequent of the UCDs.
Unlike fats and carbohydrates, protein is not stored in the body but rather exists in a balanced state of anabolism (formation) and catabolism (breakdown). Protein excess beyond normal bodily requirements comes from either excess dietary protein intake or from protein breakdown through various catabolic processes (stress of the newborn period, infection, dehydration, injury, or surgery). Amino acids liberated from excess protein are broken down, releasing nitrogen which circulates in the body as ammonia (NH3). Ammonia is then converted into urea via the urea cycle and disposed of in the urine. An enzymatic block in the urea cycle defect results in the accumulation of excess ammonia which has toxic effects, most severe in the central nervous system causing cerebral edema.
Ammonia also circulates in the body as free ammonia or within glutamine which functions as a temporary “repository” for ammonia. Consequently, in a urea cycle defect not only does free ammonia rise (hyperammonemia) but glutamine is also elevated. Alanine (Ala) is another amino acid that accumulates as a result of hyperammonemia due to a urea cycle defect. These two amino acid elevations (glutamine, alanine) may precede hyperammonemia and the onset of clinical symptoms and can serve as useful biochemical markers of decompensation in a patient with a urea cycle defect
Clinical Presentation of an Acute Hyperammonemic Episode
Poor feeding
Lethargy
Tachypnea
Hypothermia
Irritability
Vomiting
Ataxia
Seizures
Hepatomegaly
Coma
Hyperammonemic crises in neonates or infants with CPS I deficiency are frequently precipitated by sepsis. Consequently, any neonate or young infant with CPS I deficiency and clinical signs of a severe illness should be evaluated for a hyperammonemic crisis precipitated by infection or any stressor.
Clinical Assessment
Assess cardiac, respiratory, neurologic, and hydration status
Identify potential precipitant(s) of metabolic decompensation such as infection (presence of fever) or any other physical stressor (e.g. injury, surgery)
Initial laboratory tests to order:
Plasma ammonia (1.5 ml blood in sodium-heparin tube sent STAT to lab on ice, run immediately)
Plasma amino acids
Urine orotic acid
Liver function tests (AST, ALT, alkaline phospatase, bilirubin)
Arterial or venous blood gas
Serum electrolytes, bicarbonate, BUN, creatinine
Blood glucose
Blood, urine, and/or CSF cultures (as clinically indicated)
Plasma ammonia level is a direct index of CNS toxicity and is important to follow in acute management.
Plasma amino acids should be drawn at presentation and should be monitored frequently thereafter. Glutamine, as an ammonia buffer, reflects the direction of control of the hyperammonemia and, therefore, it is a useful marker for monitoring ammonia status.
Other amino acids, including glutamate, glycine, asparagine, aspartate and lysine, may be elevated when there is an excess in waste nitrogen burden.
Please note that prolonged therapy with phenylacetate (phenylbutyrate) and/or benzoate may lead to a disproportionate decrease or only a modest elevation of glutamine and/or glycine.
Laboratory Findings in CPS I Deficiency
High plasma ammonia level
High plasma glutamine and alanine levels, low plasma citrulline level; low plasma arginine level
Absent or normal urine orotic acid level
Respiratory alkalosis (initially)
Treatment
Immediate treatment of hyperammonemia is crucial to prevent neurologic damage and avoid associated morbidity and mortality. Cognitive outcome is inversely related to the number of days of neonatal coma in the urea cycle disorders. Rapid control of the hyperammonemia is crucial in preventing or lessening the degree of mental retardation.
Key steps of immediate treatment:
Stop all protein intake (but do not withhold protein for longer than 36-48 hours as that can promote breakdown of endogenous proteins and hamper metabolic control).
Provide intravenous fluids with dextrose and intralipids: 10%-25% IV dextrose at 1.5 times maintenance rate and intralipid at 1-3 g/kg/day to provide 120-130 kcal/kg/day. This will require a central line.
Provide ammonia scavenger medications as detailed below.
Prepare for probable hemodialysis by contacting the relevant renal and surgical specialists in anticipation of imminent need.
The treatment of a hyperammonemic crisis in a patient with CPS I deficiency rests on the principles which are detailed below in the following sections:
A. Promote waste nitrogen excretion
B. Reverse catabolism/optimize caloric intake
C. Treat the underlying precipitant
A. Promote Waste Nitrogen Excretion
There are two main ways to promote ammonia detoxification: hemodialysis and medications that facilitate ammonia excretion.
Hemodialysis is the most effective way of rapidly disposing of excess ammonia and is far superior to other methods of dialysis (hemofiltration, peritoneal dialysis). Hemodialysis has the added benefit of removing amino acids such as glutamine and, in that way, disposing of additional waste nitrogen from the body A newborn or young infant with a plasma ammonia greater than 300 μmol/L, should have hemodialysis ASAP, and administer IV Ammonul until hemodialysis is instituted. Central venous catheters should be placed in a critically ill patient inhyperammonemic crisis in anticipation of the potential for hemodialysis and the appropriate nephrology and surgical specialists should be alerted in advance for this potential need. The decision to hemodialyze is critical in preventing or minimizing irreversible CNS damage; when in doubt in the face of a markedly elevated ammonia level, the decision should be to hemodialyze as soon as possible.
Ammonia scavenger medications include IV Ammonul® (sodium benzoate and sodium phenylacetate). Sodium benzoate conjugates with glycine to form hippuric acid and sodium phenylacetate conjugates with glutamine to form phenylacetylglutamine; both compounds are excreted in the urine, thereby removing the nitrogen (N) in glycine and glutamine which contribute to the hyperammonemia.
Sodium should not be provided in supplemental IV fluids when IV Ammonul® is given since this solution contains sufficient amounts of sodium. Otherwise, hypernatremia may result.
Side effects of IV Ammonul® may occur in children, including nausea and vomiting. This may be controlled with antiemetic medications such as ondansetron, either prior to or during the infusion. Overdoses (3-5x the recommended dose) of IV Ammonul® can lead to agitation, confusion and hyperventilation. Caution should be exercised to avoid overdosing.
IV Ammonul® dosing:
0-20 kg: 2.5 mL/kg (prior to mixture with dextrose solution) IV bolus over 90 min followed by the same dose as a 24-hour infusion.
>20 kg: 55 mL/m2 (prior to mixture with dextrose solution) IV bolus over 90 min followed by the same dose as a 24-hour infusion.
IV Arginine (600 mg/kg/day) – used to provide supplemental arginine which may be deficient due to early enzymatic blocks and to stimulate the urea cycle. However, do not use if arginase deficiency has been diagnosed, in which the arginine level is elevated. After the diagnoses of citrullinemia and argininosuccinic aciduria have been ruled out, the dose may be reduced to 250 mg/kg/day.
Citrulline – used in CPS I deficiency when the child is able to have enteral feeding. Citrulline (by mouth, NG or g-tube) may help pull aspartate into the urea cycle and, thus, increase nitrogen clearance.
B. Reverse Catabolism/Optimize Caloric Intake
*Diet should be planned in conjunction with a metabolic dietitian*
The goal is to limit protein intake and prevent/reverse catabolism by providing adequate non-protein calories. All protein should be temporarily stopped. The caloric intake for neonates and infants with CPS I deficiency in a hyperammonemic crisis should be kept at least at 120-130 kcal/kg/day and at least 125% of maintenance calorie needs in older patients. Strict intake and output records should be monitored.
The caloric intake on day 1 is provided by intravenous dextrose and supplemented with intralipid to meet calorie goals. Enteral feeds (oral or NG/NJ) should be started as soon as practical and that can even occur concomitant with IV nutrition and fluids if necessary.
Ideally, protein will be reintroduced within 24-48 hours to avoid breakdown of endogenous proteins; however, this may be delayed depending on clinical status. Start protein at 0.5 grams/kg/day, administered as essential amino acids (e.g., Cyclinex-1, WND-1, etc.). After 24-48 hours of this regimen and if well tolerated, dietary protein can be gradually increased by 0.25 - 0.5 gram/kg/day to 1.2 grams/kg/day – 50% from essential amino acids and 50% from a natural protein source (e.g., regular infant formula or breast milk). Avoid elemental formulas containing all amino acids, as they tend to have a high nitrogen content. Supplemental calories are added from a protein-free formula with vitamins and minerals (e.g., Pro-Phree or equivalent). Thereafter, the protein intake can be gradually increased, as tolerated, to a maximum of 2 grams/kg/day. The rate of protein advancement and final protein goal should be tailored to the patient.
C. Treat the Underlying Precipitant
A metabolic decompensation in a patient with a UCD is often precipitated by an underlying illness such as an infection or dehydration which results in a state of catabolism. Diagnostic investigation and treatment aimed at this underlying precipitant is extremely important to optimize metabolic control of the decompensation and should be undertaken at the time of initial presentation and continued throughout the management phase of hyperammonemia.
*Caution: Do not perform lumbar puncture (LP) before evaluating for the presence of cerebral edema, which may contraindicate an LP.
Monitoring
Plasma ammonia levels do not always directly correlate with the presence or severity of clinical signs and symptoms and, thus, monitoring of clinical status and changes in that is crucial. Clinical decisions on appropriate treatments should be based on the combination of clinical assessment and plasma ammonia levels.
Monitor ammonia levels every 4 hours, and electrolytes and arterial/venous blood gas as clinically indicated. Plasma amino acids should be monitored frequently depending on levels. If another IV is required, that solution should not contain sodium if given simultaneously with IV Ammonul®
There may be a “rebound” hyperammonemia initially as stored glutamine is metabolized to glutamate and ammonia, and with the efflux of intracellular ammonia into the ‘relatively’ ammonia-depleted blood while on treatment and, thus, it is important to continue closely monitoring plasma ammonia levels until they remain stable in the normal range. On rare occasions it may be necessary to assess the magnitude of glutamine excess in brain tissue by performing brain magnetic resonance spectroscopy (MRS).
Plasma glucose levels should be kept below 150 mg%. If hyperglycemia occurs while IV 10%-20% dextrose is supplied for added calories, an IV insulin drip at 0.01 units/kg/hour should be started to maintain plasma glucose between 100 and 150 mg%. Insulin may be increased by 0.01-0.03 units/kg/hour until desired effect is obtained. Please note that as the patient’s condition improves and anabolic homeostasis is restored, it may be necessary to rapidly eliminate or reduce the rate of the insulin infusion as hypoglycemia may develop. High IV dextrose solutions should not be decreased or stopped in the face of hyperglycemia. The goal is to keep the level from rising above 150 mg/dl. Wide swings in glucose levels are not ideal and so plasma glucose should be kept within the above as best as possible.
Cerebral edema: Oncotic agents such as albumin will increase the overall nitrogen load but may in selected cases be considered. Mannitol has been used but may not be as effective as hypertonic saline in alleviating cerebral edema due to hyperammonemia. Steroids should never be used in a patient with hyperammonemia. Hyperventilation is recommended, but only under close supervision.
Neurologic status should be closely monitored for signs of CNS toxicity and cerebral edema while the patient is under treatment and in the recovery period.
Caloric intake should be kept at a high level at all times to prevent catabolism. Close monitoring of the intake of calories is an essential part of treatment and monitoring of a patient with a urea cycle defect in crisis.
Avoid certain medications, such as valproic acid, as it interferes with urea cycle function and accentuates hyperammonemia.
Recovery
Once the patient is stabilized and improving, oral diet has been established, and the plasma ammonia level is stable in an acceptable range, oral scavenger medications (sodium benzoate, sodium phenylbutyrate) and oral arginine can be provided in place of their IV forms.
The dose of sodium benzoate and sodium phenylbutyrate is determined based on either body weight or body surface area. The dose should be decided in conjunction with a metabolic physician if the patient does not have an up to date regimen.
References
The urea cycle disorders conference group. Consensus statement from a conference for the management of patients with urea cycle disorders. Supplement to the Journal of Pediatrics. Vol. 138 (1), Jan 2001.
Saudubray JM, Van den Berghe G, Walter JH. Inborn Metabolic Diseases: chapter 20, Disorders of the Urea Cycle and Related Enzymes. pp. 298-310. 2012.